
Blog
What is Thalassemia
Introduction
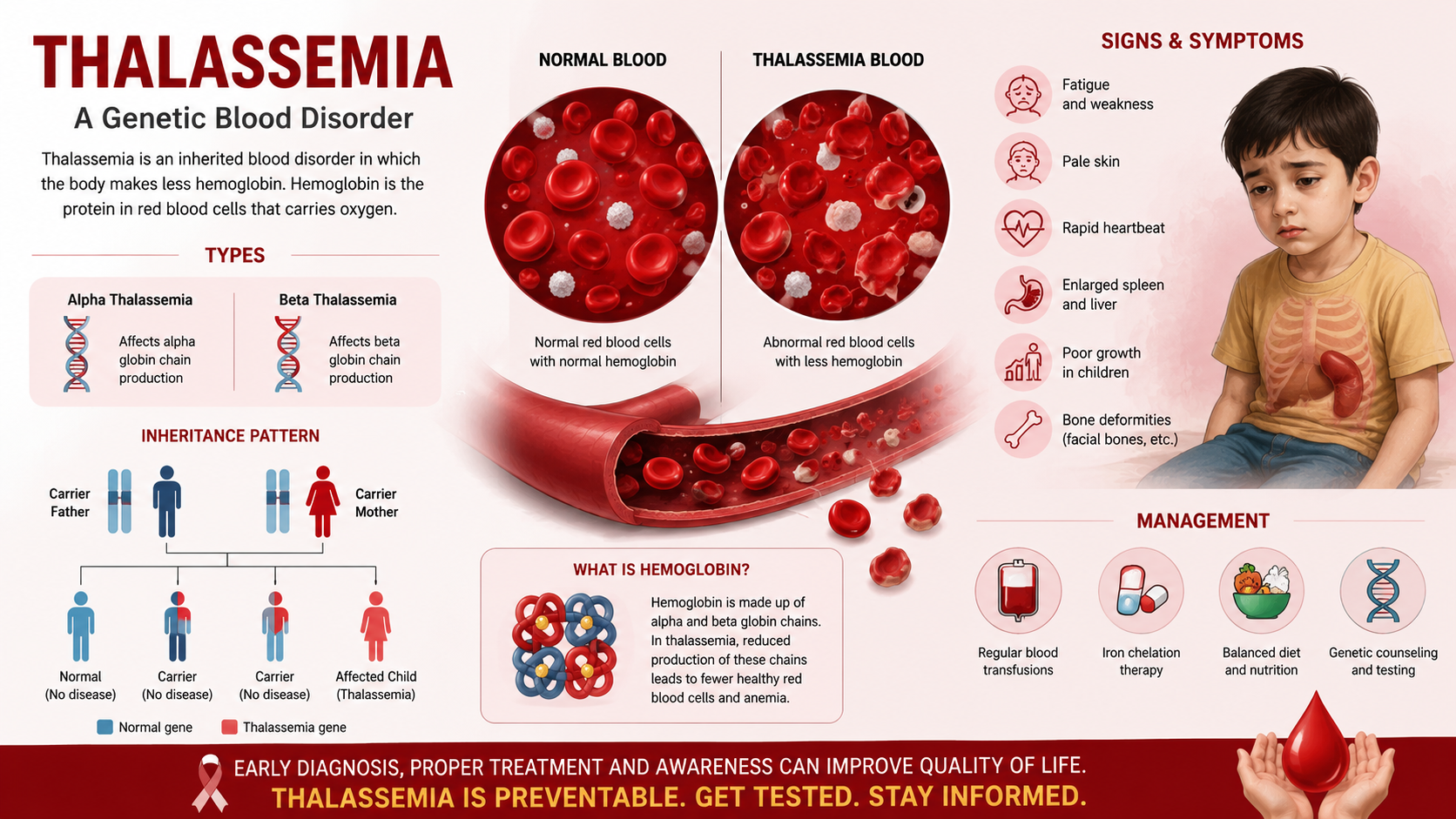
Thalassemia is a genetic blood disorder that affects the body’s ability to produce healthy hemoglobin. Hemoglobin is an important protein in red blood cells that carries oxygen from the lungs to the rest of the body. When hemoglobin production is low or abnormal, red blood cells are destroyed easily, leading to anemia.
Thalassemia is inherited from parents to children and is not caused by infection or lifestyle. People with thalassemia may experience symptoms such as fatigue, weakness, pale skin, shortness of breath, and slow growth in children. In severe cases, regular blood transfusions and medical care are required.
Causes of thalassemia:
1. Genetic Inheritance from Parents
Thalassemia is mainly caused by genetic inheritance, which means it is passed from parents to their children through genes. Genes carry instructions for making hemoglobin in the body. When parents carry defective thalassemia genes, these faulty genes can be transferred to their children. If both parents are carriers, there is a strong chance that the child will be born with thalassemia. Even if parents appear healthy, they may still carry the thalassemia gene without showing symptoms. This inherited problem affects the normal production of hemoglobin and leads to anemia.
2. Mutation in Hemoglobin Genes
Another important cause of thalassemia is mutation in hemoglobin-producing genes. A mutation is a permanent change in the genetic structure. These changes stop the body from producing normal hemoglobin. As a result, red blood cells become weak, break easily, and cannot carry enough oxygen. The severity of thalassemia depends on how serious the mutation is. Some mutations cause mild anemia, while others lead to severe thalassemia that requires lifelong treatment.
3. Family History and Consanguineous Marriages
Family history plays a major role in thalassemia. If thalassemia exists in a family, future generations have a higher risk of developing the disease. The risk increases further in consanguineous marriages (marriage between close relatives). In such cases, both parents are more likely to carry the same defective gene, increasing the chances of children being affected by severe thalassemia.
4. Regional and Ethnic Genetic Patterns
Thalassemia is more common in certain regions such as South Asia, the Middle East, Africa, and Mediterranean countries. People living in or originating from these areas have a higher chance of carrying thalassemia genes due to long-standing genetic patterns. Over many generations, these genes have remained common in these populations. This is why thalassemia is more frequently diagnosed in these regions compared to others.
Important Note: Thalassemia is not caused by poor nutrition, infections, or unhealthy lifestyle choices. It is a genetic condition and cannot spread from one person to another. Early screening, blood tests before marriage, and genetic counseling can help prevent severe forms of thalassemia and reduce its impact on families.
Genetic Inharitance of Thalassemia;
Imagine your body as a factory and genes as the instruction manual that tells the factory how to work. One very important instruction is how to make hemoglobin, the protein that carries oxygen in your blood. In thalassemia, this instruction manual has some printing mistakes. Because of these mistakes, the factory cannot make healthy hemoglobin, and this problem is passed from parents to children.
Each person receives two copies of genes—one from the mother and one from the father. Think of it like getting two recipe books. If one recipe book has a mistake and the other is correct, the dish still turns out okay. This person is called a carrier. Carriers usually look healthy and may never know they carry the thalassemia gene, but they can still pass the faulty recipe to their children.
Now imagine both parents have faulty recipe books. When they have a child, nature makes a random choice:
Sometimes the child gets two correct recipesthe child is normal.
Sometimes the child gets one correct and one faulty recipe → the child becomes a carrier.
Sometimes the child gets two faulty recipes the child develops thalassemia major, a serious condition that needs lifelong treatment.
This is why doctors say there is a 25% risk of severe thalassemia when both parents are carriers.
There are two main genetic stories in thalassemia;
In alpha thalassemia, the problem is with the genes that make alpha globin. the more missing genes, the more serious the disease.
In beta thalassemia, the beta globin genes are affected. One faulty gene causes mild disease, while two faulty genes cause severe thalassemia.
Marriages between close relatives increase the risk because family members often carry the same genetic mistakes. It is like copying the same misprinted book again and again.
The important thing to remember is that thalassemia is not a curse, infection, or weakness. It is simply a genetic condition. With awareness, blood tests before marriage, and genetic counseling, families can stop severe thalassemia before it starts and give future generations a healthier life.
Types of thalassemia:
1. Alpha Thalassemia
Alpha thalassemia occurs when the genes responsible for making alpha globin chains are missing or damaged. Normally, a person has four alpha-globin genes (two from each parent). The severity depends on how many genes are affected.
a) Silent Carrier (1 gene missing)
Only one alpha gene is defective.
The person has no symptoms and appears completely healthy.
Usually discovered only through special blood tests.
b) Alpha Thalassemia Trait (2 genes missing)
Two alpha genes are missing.
Causes mild anemia.
Person may feel tired sometimes but usually lives a normal life.
c) Hemoglobin H Disease (3 genes missing)
Three alpha genes are missing.
Causes moderate to severe anemia.
Symptoms include weakness, pale skin, enlarged spleen, and fatigue.
May require medical treatment and occasional blood transfusions.
d) Hydrops Fetalis (4 genes missing)
All four alpha genes are missing.
This is the most severe form of alpha thalassemia.
The baby usually does not survive before or shortly after birth.
2. Beta Thalassemia
Beta thalassemia occurs due to defects in the genes that make beta globin chains. Each person normally has two beta-globin genes, one from each parent.
a) Beta Thalassemia Minor (Trait)
One beta gene is defective.
Person is a carrier.
Symptoms are very mild or absent.
Mild anemia may be present, but life is usually normal.
b) Beta Thalassemia Intermedia
Both beta genes are affected, but not completely damaged.
Causes moderate anemia.
Symptoms appear later in childhood or adolescence.
May need occasional blood transfusions.
c) Beta Thalassemia Major (Cooley’s Anemia)
Both beta genes are severely defective.
Causes severe anemia in early childhood.
Children need regular blood transfusions and lifelong medical care.
Without treatment, it can be life-threatening.
3. Thalassemia Based on Severity
Thalassemia can also be grouped by severity:
Thalassemia Minor – Mild, carrier state
Thalassemia Intermedia – Moderate symptoms
Thalassemia Major – Severe, requires regular
Symptoms of Talassemia
1. Severe Anemia and Extreme Weakness
Anemia is the most common and serious symptom of thalassemia. Due to low hemoglobin levels, red blood cells cannot carry enough oxygen to the body’s organs and tissues. This leads to constant tiredness, weakness, shortness of breath, dizziness, and fast heartbeat. Children may feel exhausted even after light activity and may prefer resting instead of playing. In severe cases, anemia becomes life-threatening and requires regular blood transfusions to maintain hemoglobin levels.
2. Pale or Yellowish Skin and Eyes
People with thalassemia often have pale skin because of reduced red blood cells. In many cases, the skin and the whites of the eyes become yellow, a condition known as jaundice. This happens because abnormal red blood cells break down quickly, releasing bilirubin into the blood. High bilirubin levels cause yellowing of the skin and eyes and can also lead to dark-colored urine.
3. Delayed Growth and Physical Development
Children suffering from thalassemia, especially thalassemia major, often show slow growth and delayed puberty. Poor oxygen supply, frequent illness, and nutritional deficiencies prevent the body from growing normally. These children may be shorter than their peers and experience delayed sexual development. If not treated properly, growth delays can become permanent.
4. Bone Changes and Enlarged Organs
To compensate for anemia, the bone marrow tries to produce more red blood cells. This causes the bones, especially of the face, skull, and jaw, to expand and change shape, leading to bone deformities. At the same time, organs like the spleen and liver become enlarged because they work overtime to destroy abnormal blood cells. Enlarged organs can cause abdominal pain, swelling, and increased risk of infections.
Diagnosis of Talassemia;
Medical History and Physical Examination in Thalassemia :
Medical history and physical examination are the first and most important steps in diagnosing thalassemia. They help doctors suspect the disease even before laboratory tests are done.
During medical history, the doctor asks detailed questions about the patient’s symptoms. These include long-lasting tiredness, weakness, shortness of breath, dizziness, pale skin, yellowing of the eyes, repeated infections, and poor appetite. In children, special attention is given to slow growth, delayed puberty, and poor weight gain. The doctor also asks when the symptoms started, because thalassemia major usually shows symptoms in early childhood.
Family history is a very important part of medical history. Since thalassemia is a genetic disease, doctors ask if anyone in the family has anemia, needs regular blood transfusions, or has been diagnosed with thalassemia. Questions about marriage between close relatives are also asked, as this increases the risk of thalassemia. Information about previous children with anemia or early death in the family can strongly suggest thalassemia.
The doctor may also ask about previous treatments, such as iron supplements or blood transfusions. If anemia does not improve with iron therapy, it raises suspicion of thalassemia rather than iron deficiency anemia
During the physical examination, the doctor carefully examines the patient for visible signs of thalassemia. The skin and eyes are checked for paleness and jaundice, which indicate anemia and increased breakdown of red blood cells. The doctor examines the abdomen to check for enlargement of the spleen and liver, which is common in thalassemia due to excessive destruction of abnormal red blood cells.
Complete Blood Count (CBC) Test in Thalassemia;
The Complete Blood Count (CBC) test is one of the most important and commonly used tests to diagnose thalassemia. It gives detailed information about the blood and helps doctors detect anemia and other blood disorders.
A CBC test measures several important components of blood. One key value is hemoglobin level. In thalassemia, hemoglobin is usually lower than normal, which explains symptoms like weakness, tiredness, and shortness of breath. The test also measures the red blood cell (RBC) count. In thalassemia, the number of red blood cells may be normal or even higher than normal, but these cells are usually abnormal and do not work properly.
Another important part of the CBC test is the measurement of red blood cell size, called Mean Corpuscular Volume (MCV). In thalassemia, red blood cells are smaller than normal, a condition known as microcytosis. The test also measures Mean Corpuscular Hemoglobin (MCH), which shows how much hemoglobin is present in each red blood cell. In thalassemia, MCH is low, meaning the cells are paler and carry less oxygen.
The CBC test also provides information about white blood cells and platelets. These values are usually normal in thalassemia, but they may change if there is infection or an enlarged spleen. A high red blood cell count with low hemoglobin and small-sized cells strongly suggests thalassemia, especially when anemia does not improve with iron treatment.
Genetic Testing in Thalassemia
Genetic testing is an important method used to confirm thalassemia and to identify whether a person is a carrier or affected by the disease. Since thalassemia is a genetic disorder, this test looks directly at the genes responsible for making hemoglobin.
In genetic testing, a small sample of blood, saliva, or tissue is taken from the person. This sample is examined in a laboratory to find mutations or missing genes in the alpha or beta globin genes. These mutations interfere with normal hemoglobin production and cause thalassemia. Genetic testing can clearly show whether one or both genes are defective.
This test is especially useful when blood tests like CBC or hemoglobin electrophoresis do not give clear results. It helps doctors accurately determine the type of thalassemia, such as alpha or beta thalassemia, and also the severity of the disease. Genetic testing is the most reliable way to identify thalassemia carriers, who often have no symptoms but can pass the gene to their children.
Genetic testing is also very important for family planning. Couples can be tested before marriage or pregnancy to see if they are carriers. If both parents are carriers, genetic testing can be done during pregnancy to check whether the unborn baby is affected. This helps parents make informed decisions early.
Effect of Thalassemia on the body;
Thalassemia affects the body because it reduces the production of healthy hemoglobin. Hemoglobin is responsible for carrying oxygen in the blood. When oxygen supply is low, many organs and systems of the body are affected. The effects can range from mild to severe, depending on the type of thalassemia.
One major effect of thalassemia is chronic anemia. Low hemoglobin levels mean that body tissues do not receive enough oxygen. This causes constant tiredness, weakness, shortness of breath, dizziness, and poor physical stamina. The heart has to work harder to pump oxygen-poor blood, which can lead to a fast heartbeat and, over time, heart problems.
Thalassemia also affects growth and development, especially in children. Poor oxygen supply, repeated illnesses, and nutritional deficiencies slow down physical growth. Children may be shorter than normal and experience delayed puberty. Mental development can also be affected if anemia is severe and long-lasting.
The bones are significantly affected in thalassemia. To compensate for anemia, the bone marrow works extra hard to produce more red blood cells. This causes bones, particularly in the face, skull, and jaw, to become enlarged and misshapen. Bones may also become weaker and more likely to fracture.
Thalassemia has a strong effect on internal organs such as the spleen and liver. These organs enlarge because they work overtime to destroy abnormal red blood cells. An enlarged spleen can cause abdominal pain and increase the need for blood transfusions. In severe cases, surgical removal of the spleen may be required.
Repeated blood transfusions, often needed in severe thalassemia, can cause iron overload. Excess iron gets stored in vital organs such as the heart, liver, and endocrine glands. This can lead to serious complications like heart failure, liver damage, diabetes, and hormonal problems if not properly treated with iron-chelating medicines.
The immune system can also be affected. People with thalassemia are more prone to infections, especially if the spleen is enlarged or removed. Frequent infections can further weaken the body and delay recovery.
Treatment of Thalassemia:
Regular Blood Transfusions
Regular blood transfusions are the most important treatment for patients with moderate to severe thalassemia, especially thalassemia major. Transfusions provide healthy red blood cells that contain normal hemoglobin, helping the body get enough oxygen. This treatment reduces symptoms such as extreme weakness, shortness of breath, and poor growth in children. Most patients need blood transfusions every 2 to 4 weeks throughout their lives. While transfusions greatly improve quality of life, they can cause long-term complications if not properly managed.
Gene Therapy (Advanced Treatment)
Gene therapy is a new and promising treatment for thalassemia. It works by correcting or replacing the faulty gene responsible for abnormal hemoglobin production. Although still not widely available, gene therapy has shown encouraging results in reducing or eliminating the need for blood transfusions in some patients.
Genetic Counseling and Preventive Care
Genetic counseling helps individuals and families understand thalassemia and the risk of passing it to future generations. Carrier screening before marriage, prenatal testing during pregnancy, and public awareness programs are important preventive measures. These steps help reduce the number of children born with severe thalassemia.